讲座|皮肤原发性平滑肌肉瘤1例介绍
患者,女,80岁,右面部包括2年余,渐增大,伴破溃。
大体检查:左面部肿物:灰红皮肤组织1块,大小5.7x5.0x1.6cm,表面见一隆起,大小约4.5x2.8x2.0cm,局灶见有溃疡,切面灰黄质软。
镜下表现
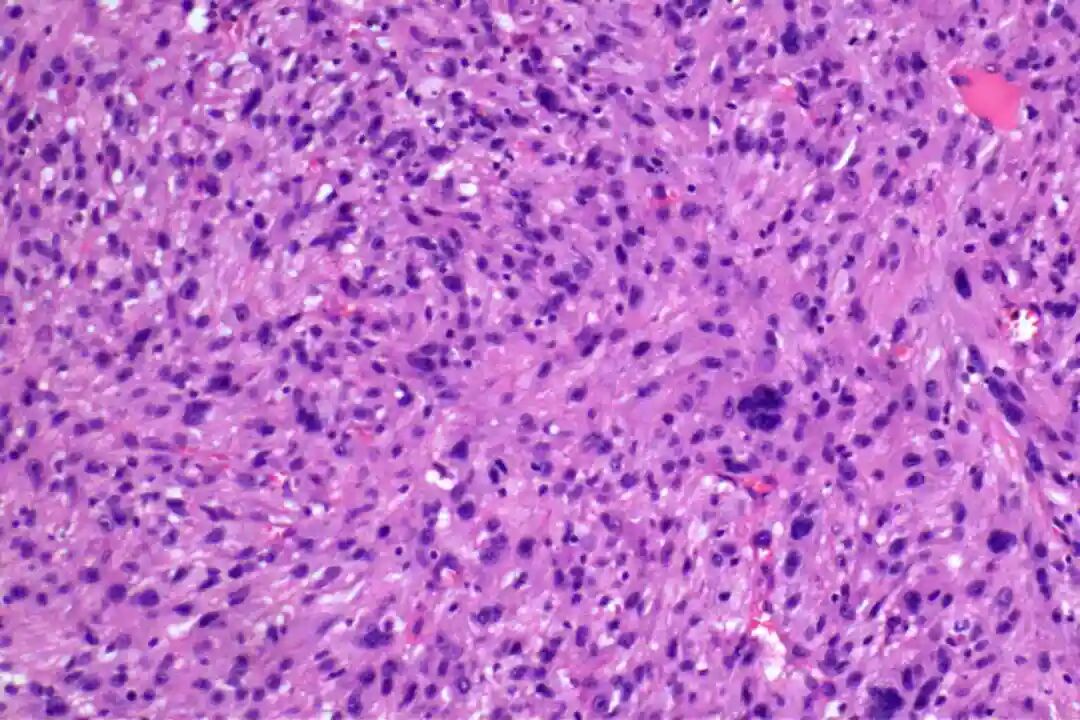
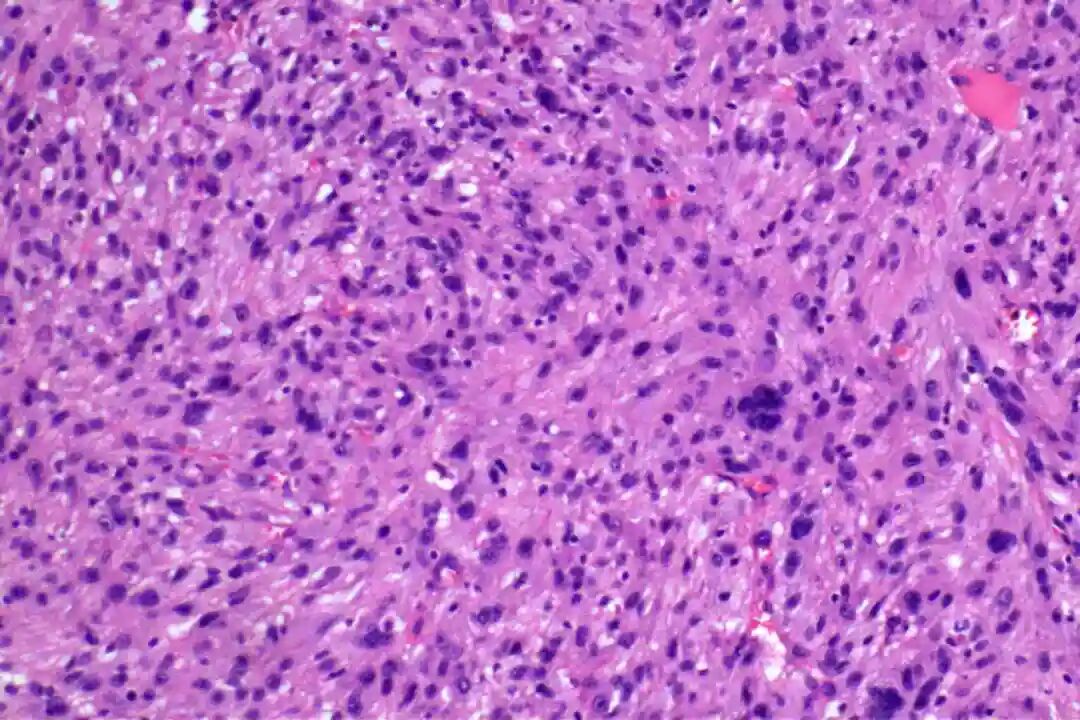
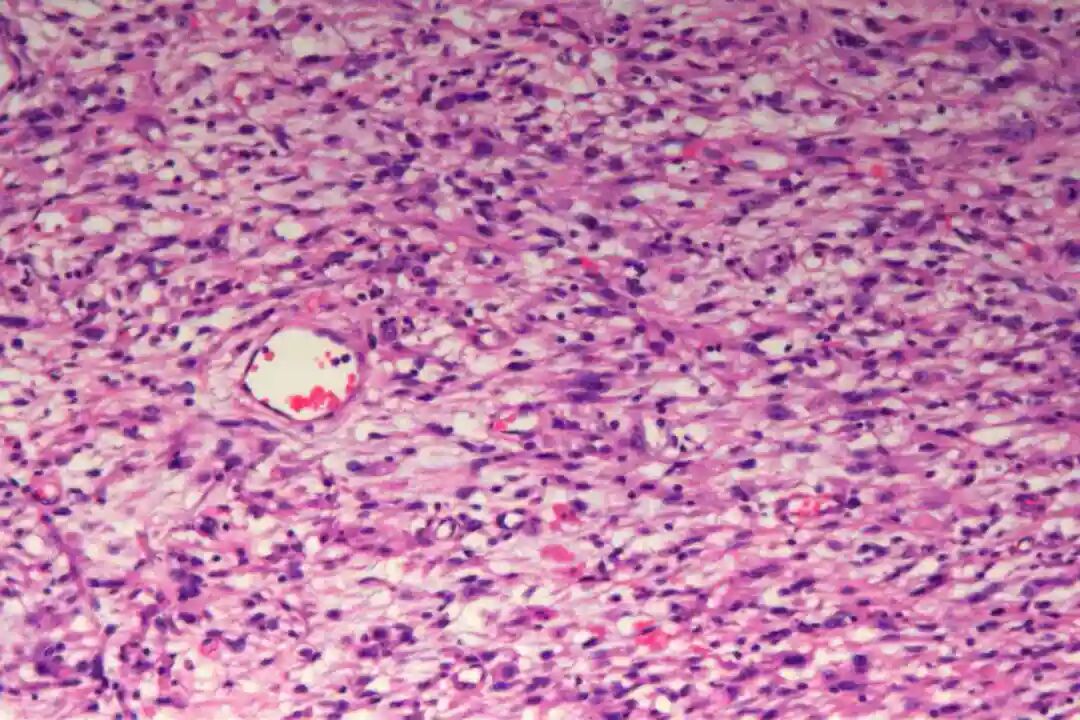
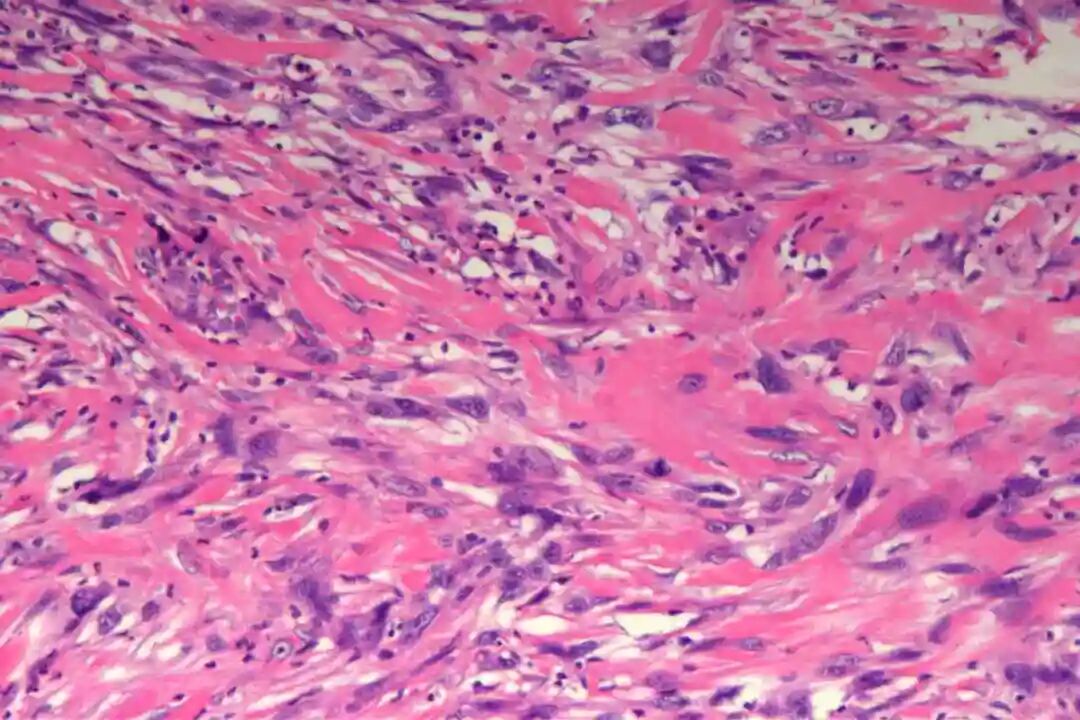
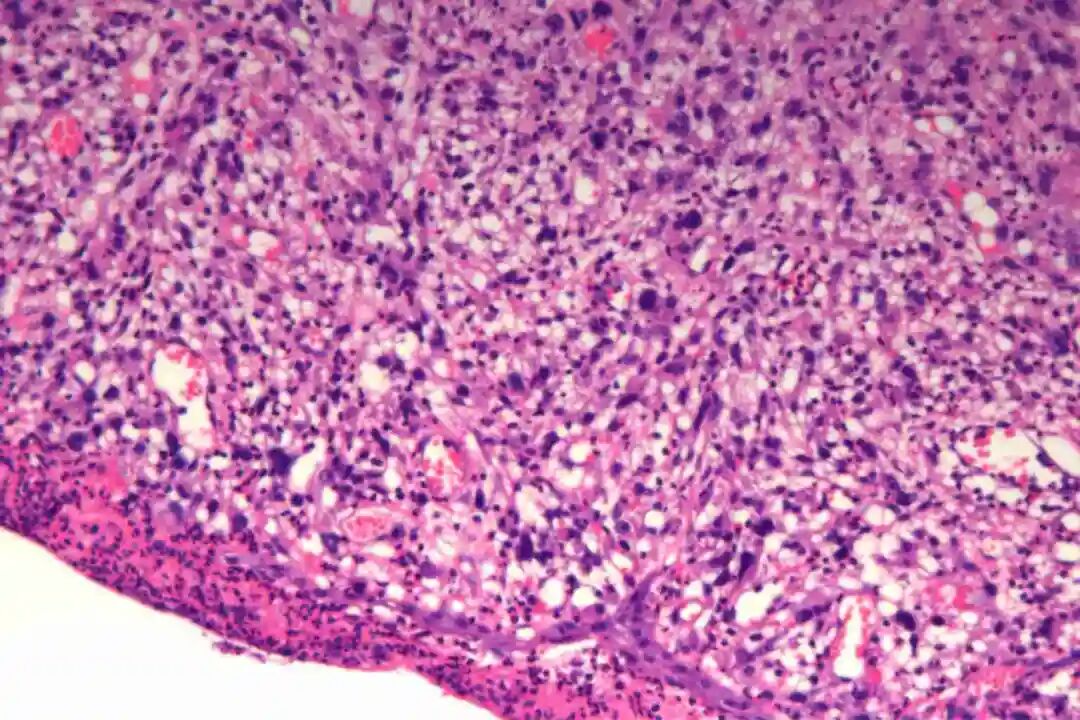
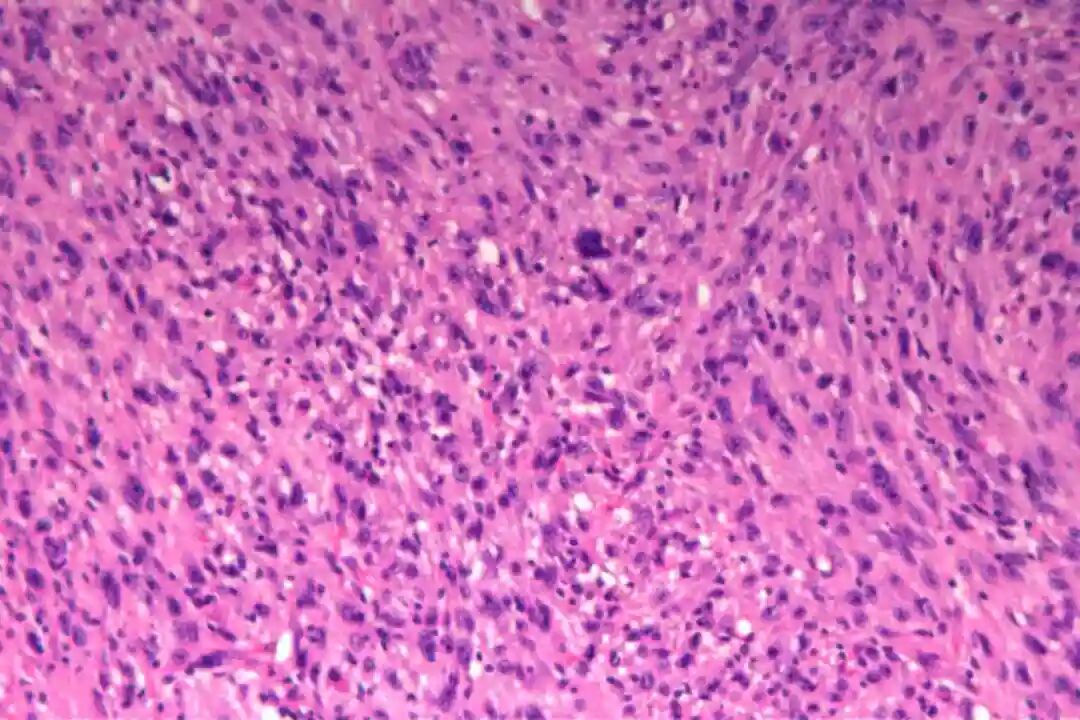
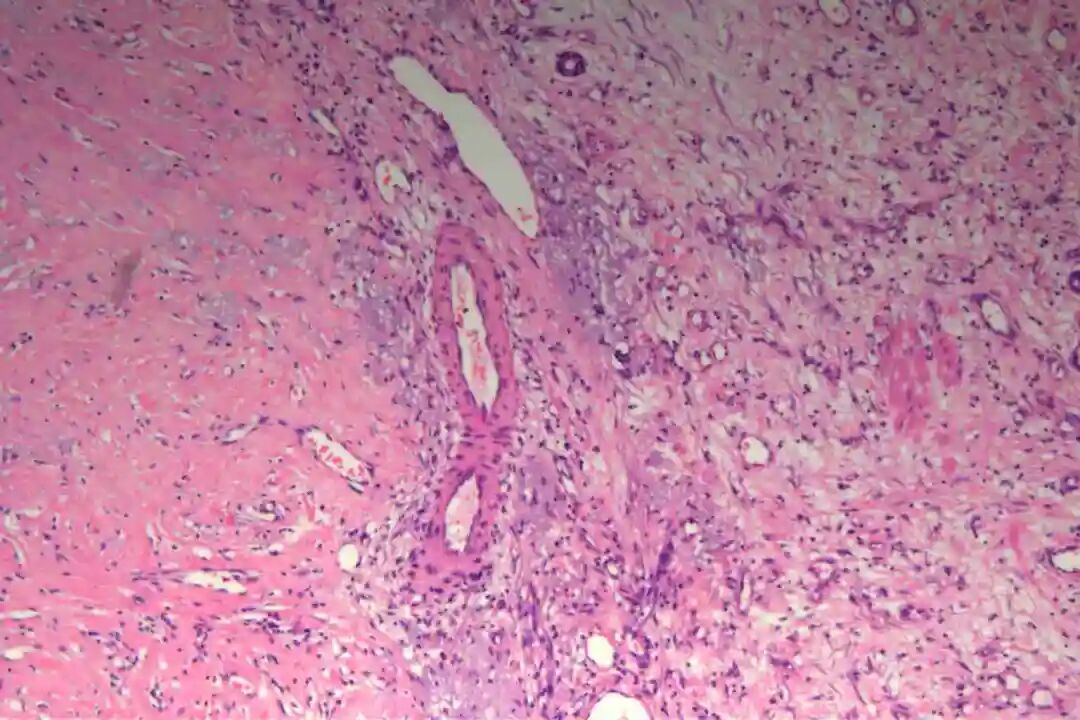
低倍镜:梭形细胞呈交织束状排列,浸润真皮深层或皮下脂肪
高倍镜:细胞核两端钝圆("雪茄形"),核异型性程度因分级而异;核分裂象(关键指标):低级别<5/10HPF,高级别≥5/10HPF;特征性胞质嗜酸性(肌源性分化)。
亚型:经典型(最常见)、黏液样型(预后较好)、上皮样型(罕见,侵袭性强);
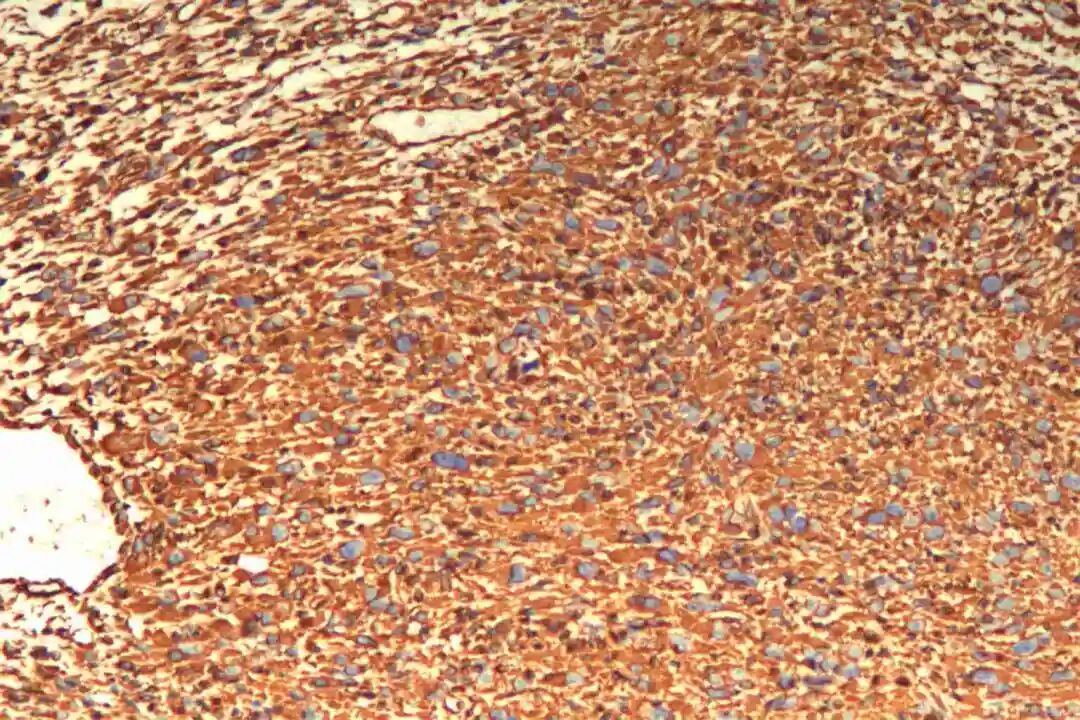
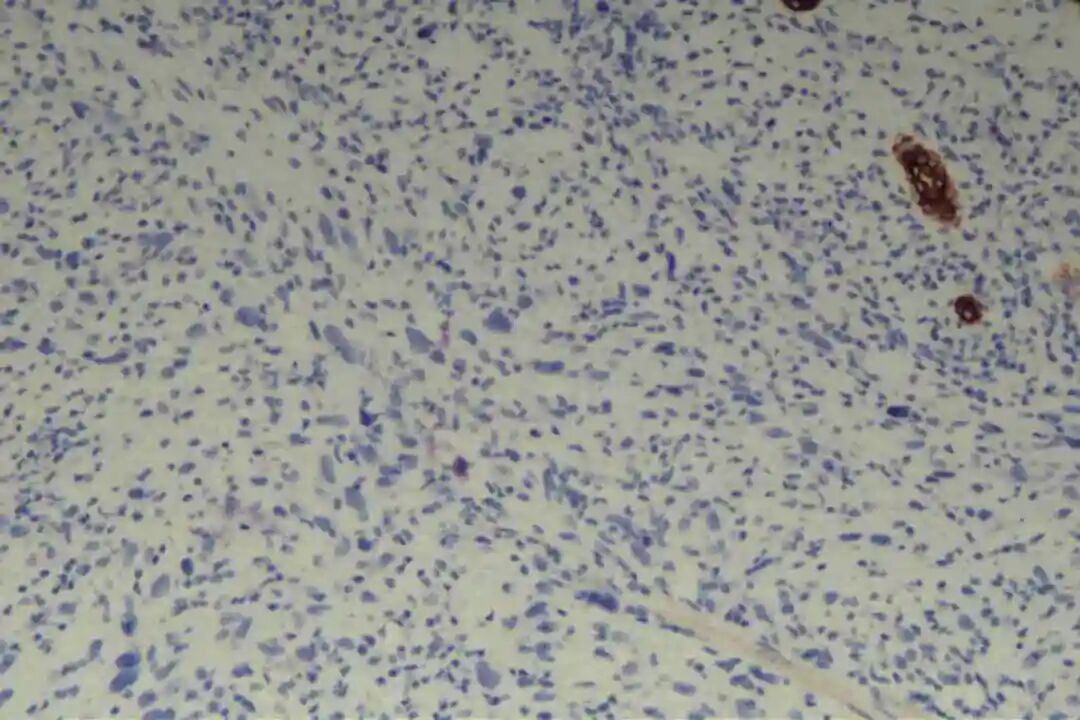
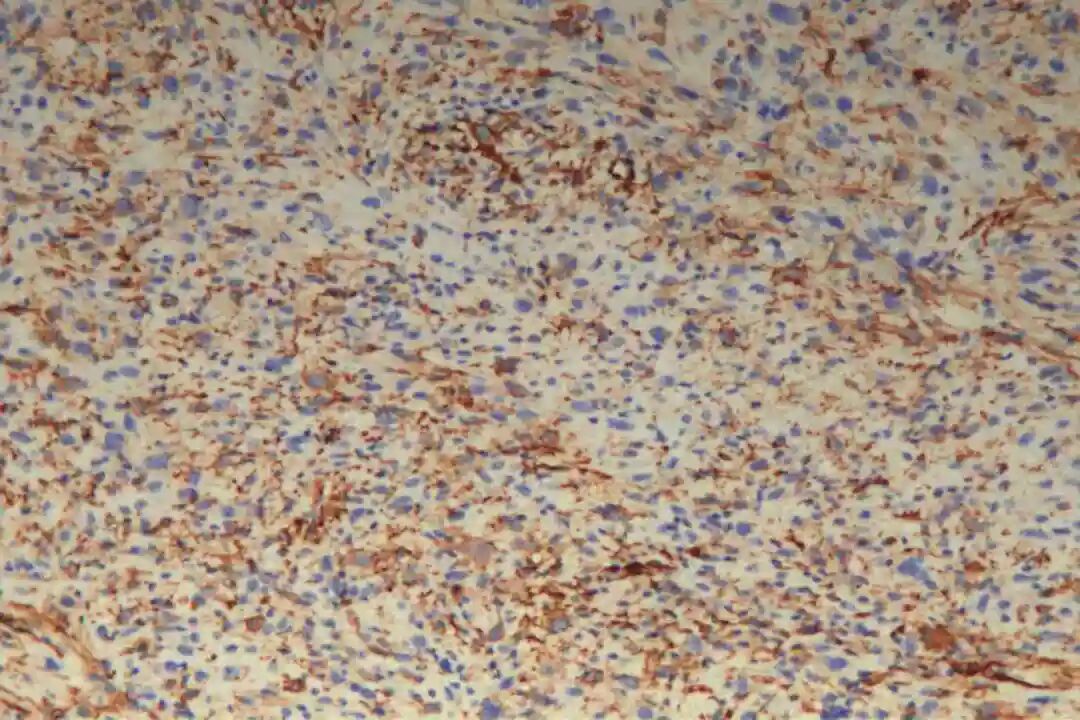
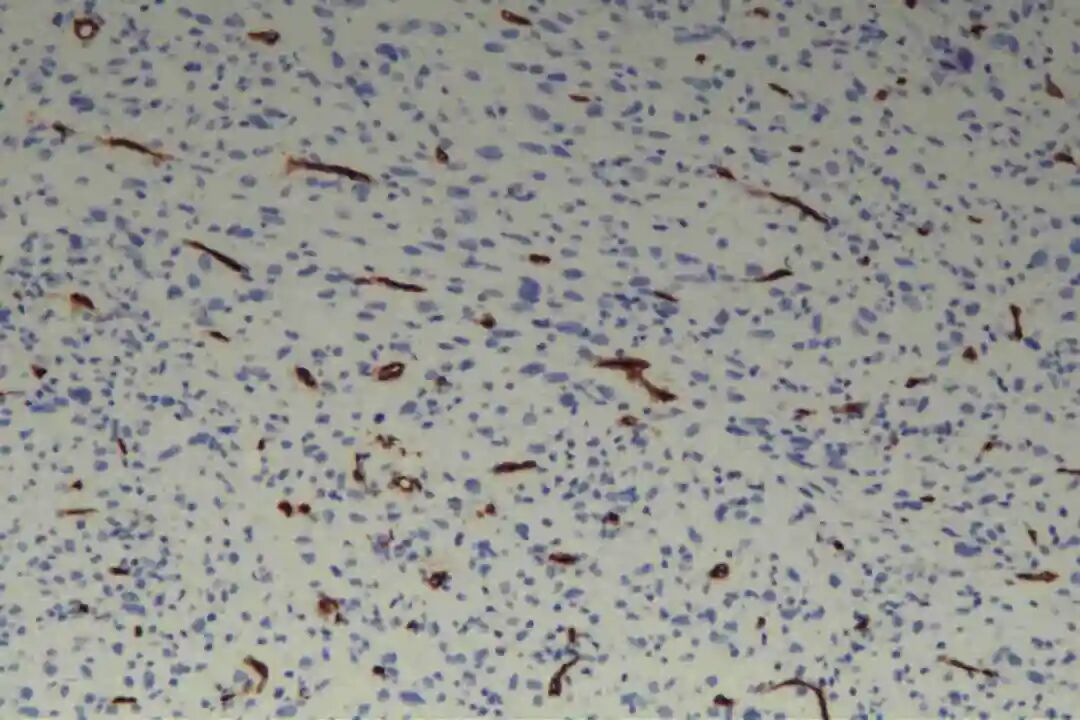
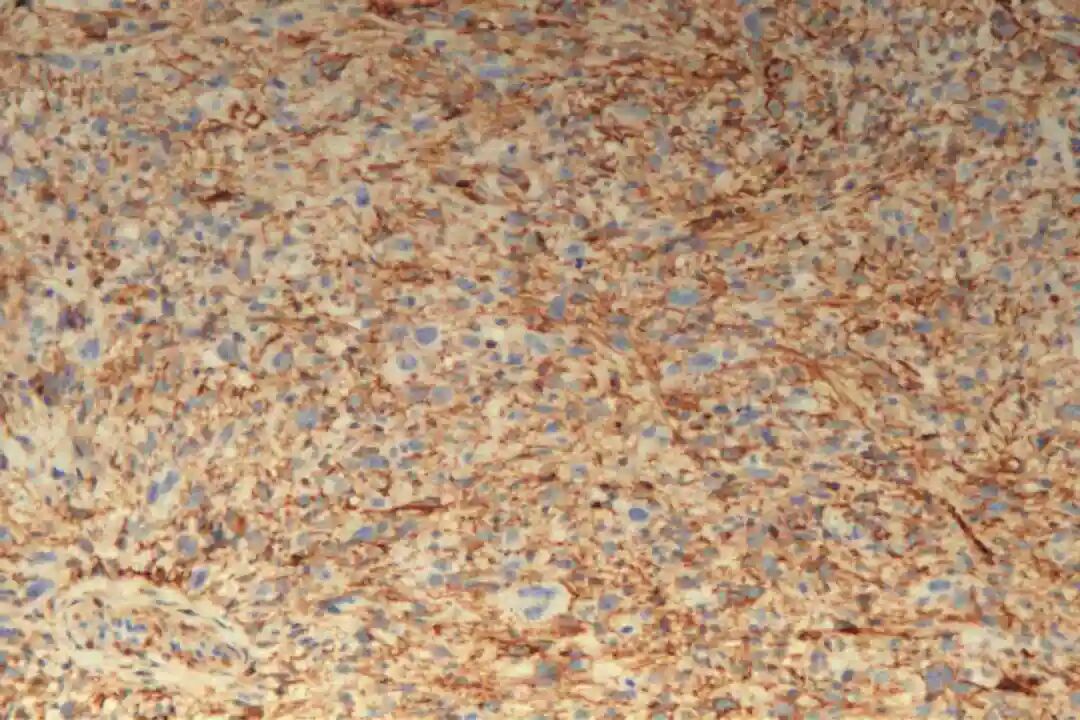
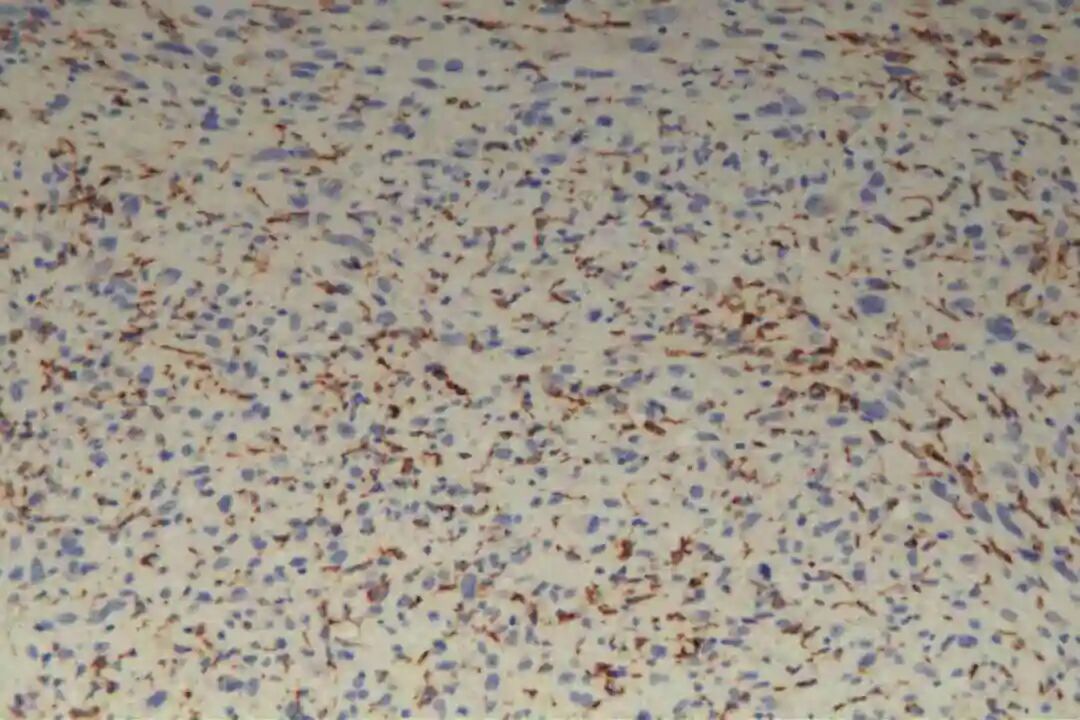
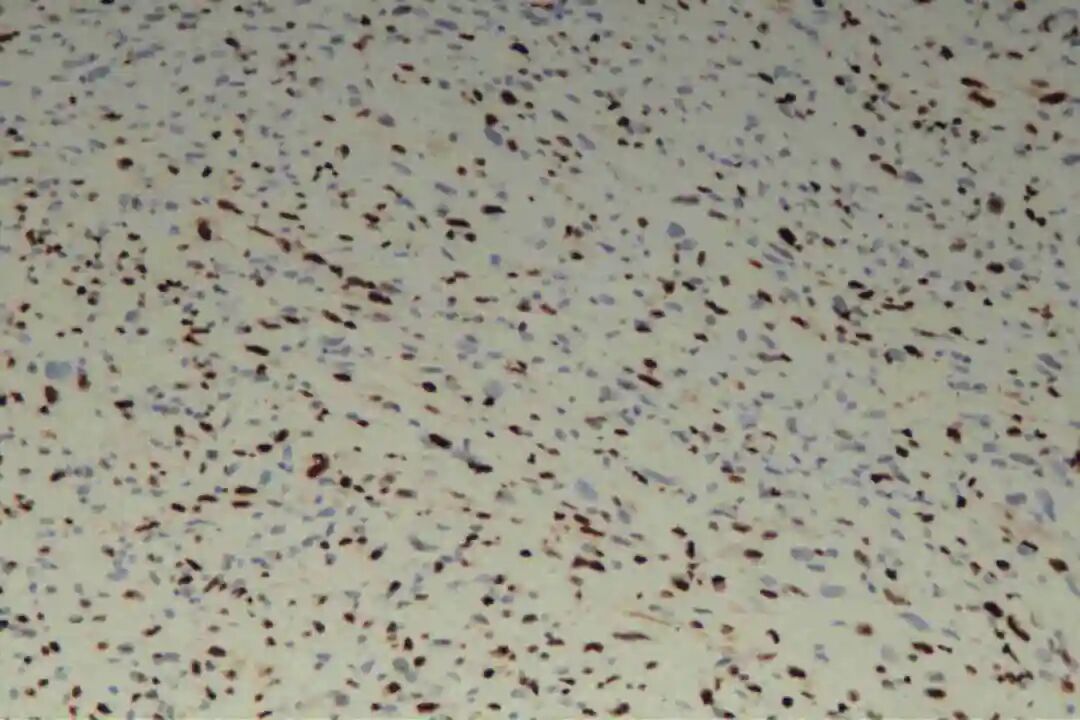
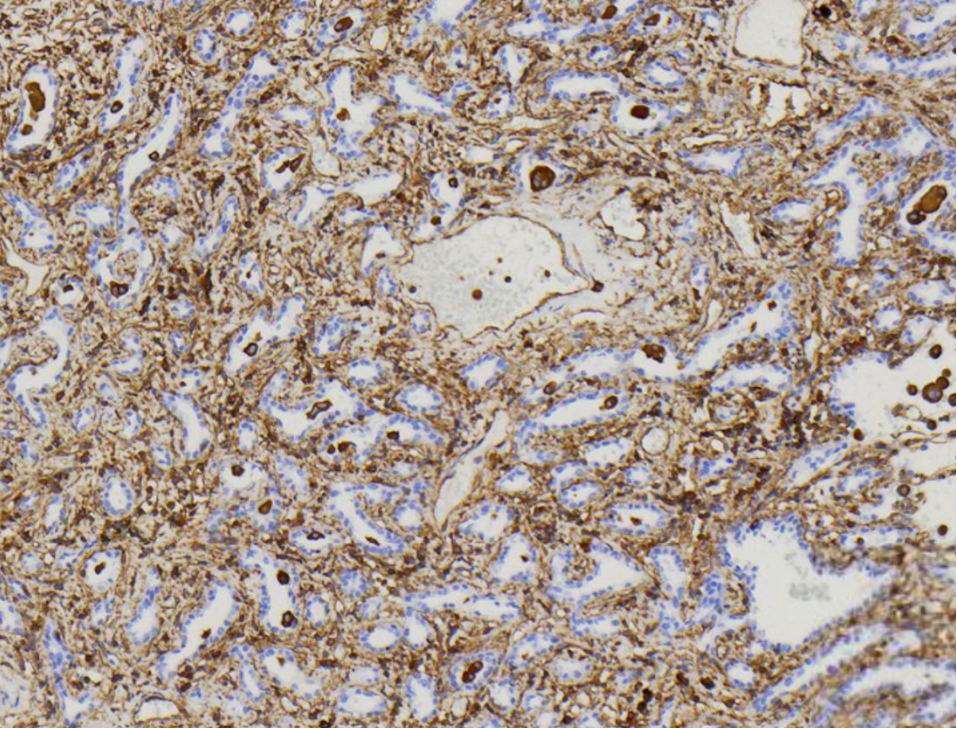
免疫组化:Vimentin(+),CKpan(-),SMA(+),Desmin(-),CD34(-),S-100(-),BCL-2(+),CD68(部分+),Ki67(40%+)。
病理诊断:(左面部)皮下梭形细胞肿瘤,结合免疫组化标记结果,符合平滑肌肉瘤,瘤体大小约4.5x2.8x2.0cm,浸润至皮下纤维脂肪组织,四周及基底切缘净;另送“左面部肿瘤残留物”未见肿瘤成分。
皮肤原发性平滑肌肉瘤(Primary Cutaneous Leiomyosarcoma,PCLMS)
一、流行病学
1、发病率:罕见,占所有软组织肉瘤<1%,皮肤肉瘤的2-3%;
2、年龄与性别:中老年多见(50-70岁),男性略多于女性(1.5:1);
3、发病部位:好发于四肢伸侧(尤其下肢)、躯干,与立毛肌或血管平滑肌相关;
4、危险因素:放射暴露史、免疫抑制(如移植后)、遗传综合征(如Li-Fraumeni综合征)。
二、临床症状
1、典型表现:单发、质硬、固定性皮下结节或斑块,直径通常1-5cm;
2、伴随症状:疼痛(50%病例,与肿瘤压迫神经或平滑肌收缩相关)、表面皮肤可呈红斑或溃疡;
3、生长特点:缓慢进展,偶见快速增大(提示高级别转化);
三、影像学特征
1、超声:不均匀低回声团块,血流信号丰富(Doppler);
2、MRI(金标准)
T1WI:等/低信号,与肌肉相似;
T2WI:高信号,伴周围水肿带;
增强:明显不均匀强化;
3、CT/PET-CT:用于评估深部浸润或转移(罕见,但高级别者可累及肺、骨);
四、病理学特征
1、大体形态:界限不清的灰白色质硬肿块,切面呈旋涡状或鱼肉样;坏死/出血可见于高级别肿瘤。
2、镜下特征
低倍镜:梭形细胞呈交织束状排列,浸润真皮深层或皮下脂肪。
高倍镜:细胞核两端钝圆("雪茄形"),核异型性程度因分级而异;
核分裂象(关键指标):低级别<5/10HPF,高级别≥5/10HPF;特征性胞质嗜酸性(肌源性分化)。
亚型:经典型(最常见)、黏液样型(预后较好)、上皮样型(罕见,侵袭性强);
3、免疫组化
必标套餐:SMA(+)、desmin(+)、h-caldesmon(+)
辅助标记:阴性:S100(排除神经源性肿瘤)、CD34(排除DFSP)、MDM2(排除ALT/WDLS);Ki-67指数(与分级相关);
新兴标记:MYOCD(平滑肌分化转录因子)
4、分子检测:无特异性突变,但常见:
基因组不稳定性(TP53、RB1缺失)
表观遗传改变(TET2、DNMT3A突变)
鉴别诊断价值:缺乏EWSR1重排(区别于EWSR1::FUS融合肉瘤);无MDM2扩增(区别于去分化脂肪肉瘤)。
五、治疗方式
1、首选广泛切除:(切缘≥2cm或筋膜切除),Mohs手术适用于面部等特殊部位;前哨淋巴结活检(争议性,仅推荐高级别或深部肿瘤)。
2、辅助治疗
放疗:用于切缘阳性或高级别肿瘤(50-60Gy);
化疗(证据有限):多柔比星+达卡巴嗪用于转移性病例;
靶向/免疫治疗:抗血管生成药(帕唑帕尼)用于晚期患者;PD-1抑制剂(如帕博利珠单抗)在TMB-H病例中试验性应用。
六、预后判断
1、关键因素
组织学分级(FNCLCC系统):高级别者5年生存率40-50% vs 低级别>90%;
肿瘤部位:头颈部预后较差(易复发);
切缘状态:R0切除者复发率<10%;
2、随访建议:每3-6个月临床检查+影像学(持续5年);肺CT(高级别者每年1次)。
七、鉴别诊断
1、良性病变
平滑肌瘤(无核分裂、无浸润)
纤维组织细胞瘤(CD34+,SMA-)
2、恶性病变
DFSP:CD34+,SMA-,COL1A1::PDGFB融合
梭形细胞黑色素瘤:S100+,SOX10+
外周神经鞘瘤:S100强阳性,SOX10+
八、最新进展(2023-2024)
1、分子分型:新发现ACTB::GLI1融合亚型(黏液样PCLMS,对SMO抑制剂敏感);
2、液体活检:ctDNA监测微小残留病(MRD)的探索性研究。